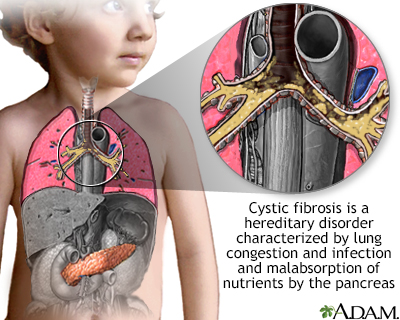
Cystic Fibrosis is a genetic disorder affecting the lungs and digestive system. Symptoms include persistent coughing, frequent lung infections, and poor growth.
Cystic Fibrosis (CF) is a serious condition that affects thousands worldwide. This genetic disorder primarily targets the lungs, leading to severe respiratory problems. Patients often experience persistent coughing and frequent lung infections. The disease also impacts the digestive system, causing malnutrition and poor growth.
Early diagnosis and treatment can improve quality of life. Understanding the symptoms is crucial for early detection and management. This article will outline key symptoms of Cystic Fibrosis, helping you recognize the signs and seek timely medical advice. Stay informed and proactive in managing this challenging condition.

What is Cystic Fibrosis?
Cystic Fibrosis is a genetic disorder affecting the lungs and digestive system. Symptoms include persistent coughing, frequent lung infections, and poor weight gain. Early diagnosis and management are crucial for improving quality of life.
Brief Overview
Cystic fibrosis is a genetic disorder. It affects the lungs and digestive system. Thick mucus builds up in the organs. This makes it hard to breathe and digest food.
The disease is inherited from parents. Both parents must carry the gene. Cystic fibrosis is more common in some populations. It can cause serious health problems.
Importance Of Awareness
Knowing about cystic fibrosis helps in early diagnosis. Early treatment can improve quality of life. People can manage symptoms better. Awareness also supports research and funding. This leads to better treatments. Families can get the support they need. Communities can help those affected by the disease.
Causes Of Cystic Fibrosis
Cystic fibrosis arises from a genetic mutation affecting the CFTR protein, leading to thick, sticky mucus in organs. Symptoms include persistent coughing, lung infections, and digestive issues.
Genetic Factors
Cystic fibrosis is caused by a defective gene. This gene is called the CFTR gene. The CFTR gene affects the cells that produce mucus, sweat, and digestive juices. With cystic fibrosis, the mucus becomes thick and sticky. This leads to severe respiratory and digestive problems. Both parents must pass on the defective gene for a child to have cystic fibrosis.
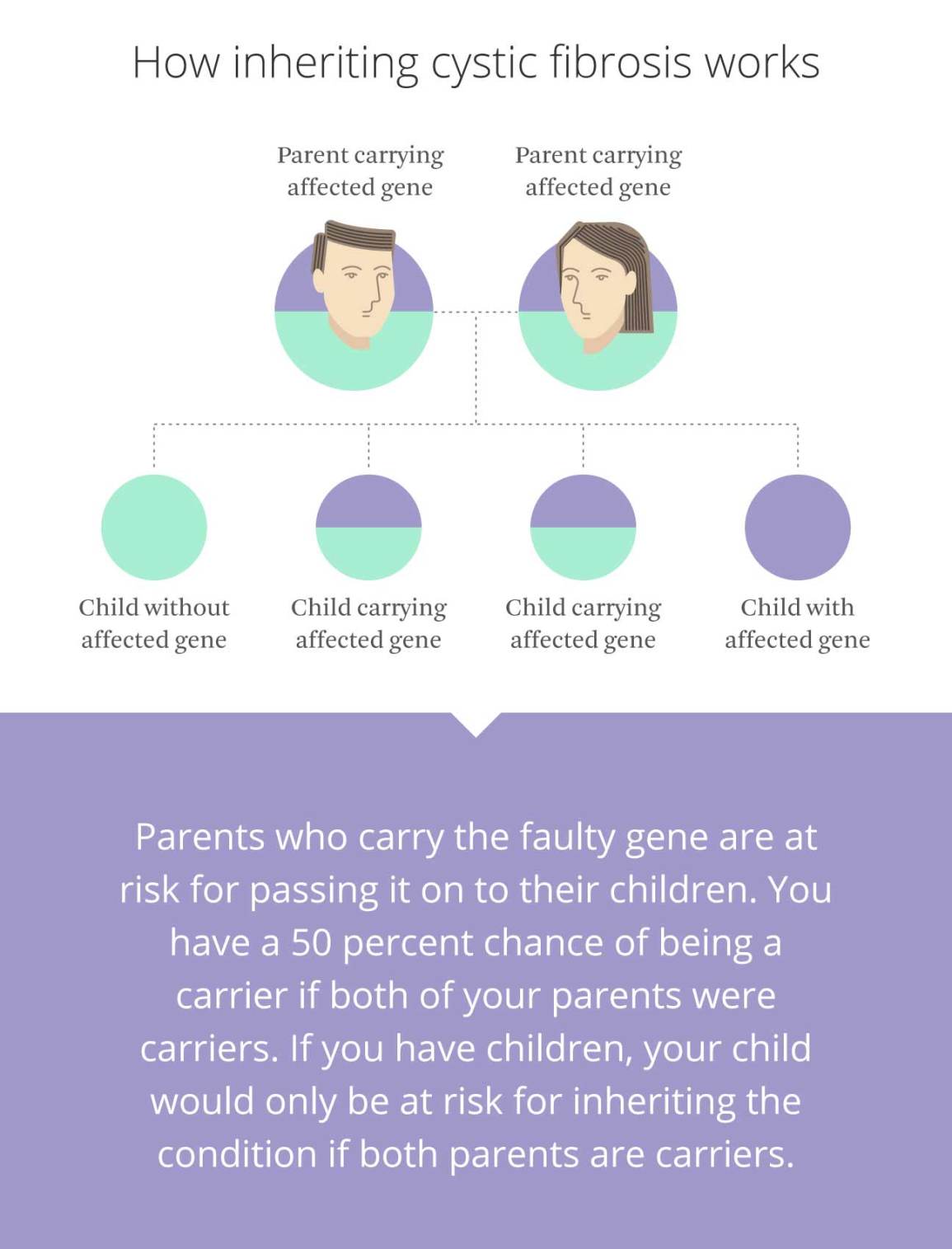
Inheritance Patterns
Cystic fibrosis follows an autosomal recessive inheritance pattern. This means a child must inherit two copies of the defective gene to have the disease. If the child gets one defective gene, they become a carrier. Carriers do not show symptoms. If both parents are carriers, each child has a 25% chance of having cystic fibrosis. There is a 50% chance the child will be a carrier. The chance of the child being unaffected and not a carrier is 25%.
Common Symptoms
People with cystic fibrosis often have thick, sticky mucus. This can clog their lungs. They may have a persistent cough. Frequent lung infections are common. Breathing may become difficult. Wheezing and shortness of breath can occur. These issues can get worse over time.
The thick mucus also affects the digestive system. It can block the release of digestive enzymes. This makes it hard to absorb nutrients. People may suffer from poor growth. Stomach pain and bloating are frequent. Greasy and bulky stools are another sign. They often have trouble gaining weight.

Lesser-known Symptoms
Cystic fibrosis often presents with lesser-known symptoms like salty-tasting skin and persistent coughing. Early identification of these signs can lead to better management.
| Reproductive Challenges | Growth Delays |
| Difficulty conceiving due to thick mucus affecting fertility. | Children with CF may experience slower growth rates. |
| Men may have absence of the vas deferens causing infertility. | Puberty can be delayed due to nutritional deficiencies. |
Diagnosis Process
Screening tests help find Cystic Fibrosis early. Newborns often get these tests. A small blood sample is taken from the baby’s heel. The sample is checked for high levels of a chemical called IRT. High IRT can mean a risk of Cystic Fibrosis. Sometimes, a second test is needed to confirm. Early detection helps start treatment sooner.
Genetic testing checks for specific gene changes. These changes cause Cystic Fibrosis. A blood or saliva sample is used. The sample is tested in a lab. If the gene changes are found, it confirms the diagnosis. Family members might also be tested. This helps find out if they carry the gene. Knowing this can help with family planning.
Treatment Options
Some medications help thin the mucus in the lungs. These make it easier to cough out the mucus. Antibiotics fight lung infections. Bronchodilators open airways and make breathing easier. There are also medicines that help the pancreas work better.
Physical therapy helps clear mucus from the lungs. Special breathing exercises make the lungs stronger. Nutritional therapy ensures the body gets needed nutrients. High-calorie meals and vitamin supplements are important.
Living With Cystic Fibrosis
Managing cystic fibrosis daily is very important. People need to take medicines. Breathing exercises help keep lungs clear. Eating healthy food is also crucial. Special diets may be needed. Regular check-ups with doctors are necessary. Family support makes a big difference. Staying active is important too. Exercise helps the body stay strong.
Support systems are vital for living with cystic fibrosis. Friends and family provide emotional support. They help with daily tasks. Support groups offer a sense of community. Meeting others with cystic fibrosis can be comforting. Medical teams offer professional help. They guide treatment and care plans. Schools and workplaces can make special accommodations. They help make life easier.
Future Outlook
Cystic fibrosis presents serious respiratory and digestive issues. Symptoms include persistent cough, lung infections, and poor weight gain. Early detection and treatment are crucial for managing this genetic disorder effectively.
Research Advances
Scientists are working hard to find new treatments. New drugs help people live longer. Gene therapy shows promise for the future. Researchers aim to fix the faulty gene causing cystic fibrosis. Clinical trials test new medicines and treatments.
Many organizations fund research to find a cure. These groups support families and patients. They also educate the public about cystic fibrosis. Advances in technology help scientists understand the disease better. New tools allow for better diagnosis and care.
Hope For A Cure
There is hope for a cure in the near future. Many people support research efforts. Donations help fund important studies. Scientists believe a cure is possible. Families remain hopeful as new treatments emerge. Every year, new discoveries bring us closer to a cure.
Living with cystic fibrosis is challenging. But advances in research bring hope. The future looks brighter for patients and their families. With continued support, a cure may soon be found.
Frequently Asked Questions
What Is Cystic Fibrosis?
Cystic fibrosis is a genetic disorder affecting the lungs and digestive system. It causes thick, sticky mucus buildup in organs, leading to respiratory and digestive issues.
What Are The Symptoms Of Cystic Fibrosis?
Symptoms include persistent cough, frequent lung infections, wheezing, shortness of breath, and poor growth. Digestive symptoms may include greasy stools and difficulty gaining weight.
How Is Cystic Fibrosis Diagnosed?
Cystic fibrosis is diagnosed through genetic testing and sweat tests. Early diagnosis is crucial for effective management and treatment.
Can Cystic Fibrosis Be Cured?
Currently, there is no cure for cystic fibrosis. However, treatments can help manage symptoms and improve quality of life.
Understanding cystic fibrosis is crucial for early detection and management. Recognizing symptoms early can improve quality of life. Stay informed and consult healthcare professionals for guidance.
Awareness and education are key in combating this condition effectively. Share this knowledge to help others understand and manage cystic fibrosis better.